Alex Rives, hoy Head of Science en BioHub, anunció ESMFold2, un motor científico abierto para alimentar predicción, diseño y descubrimiento a lo largo de la biología de proteínas.
Construido sobre datos de Cryo-EM, ESMFold2 reporta state of the art en interacciones de proteínas, especialmente anticuerpos —una modalidad crítica para terapéutica—, y evidencia de que el inference time scaling también funciona en cinco targets en cáncer e inmunología.
En un guiño a ese otro famoso proyecto de IA aplicada al protein folding, BioHub también libera un atlas de 6.800 millones de proteínas y 1.100 millones de estructuras predichas, disponible en su sitio.
¿Qué cambia respecto de AlphaFold?
Una idea que se repite en los podcasts de ciencia es que el plegado de proteínas, el diseño de materiales y la biología celular son problemas muy distintos al modelado de lenguaje. Lo son. Y sin embargo, el equipo ESM en BioHub acaba de liberar un preprint y modelo que demuestra que modelos transformer tipo BERT, entrenados sobre datasets suficientemente grandes y diversos, pueden vencer a modelos especializados como AlphaFold3 en algunos de los problemas de proteínas más difíciles.
AlphaFold2 fue revolucionario cuando se publicó en 2020: de pronto resolvía en una GPU de escritorio problemas que DESRes atacaba con clusters de supercomputación basados en ASICs custom. John Jumper y Demis Hassabis recibieron el Nobel de Química 2024 por este trabajo.
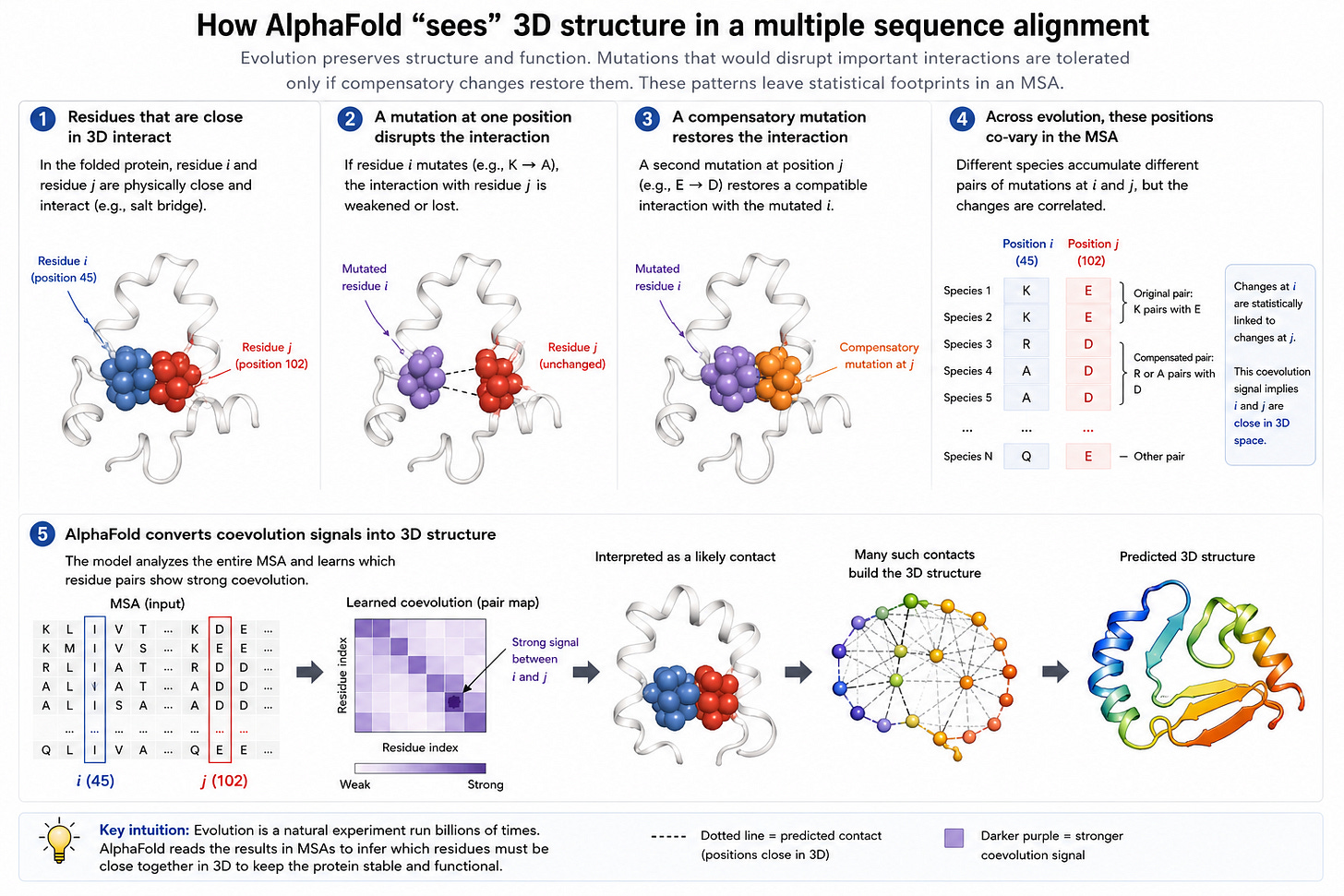
AlphaFold2 explotó una observación inteligente: si múltiples especies co-evolucionan pares de mutaciones, eso implica que las mutaciones corresponden a partes de la proteína cercanas en el espacio tridimensional. Esto se conoce como MSAs (multi-sequence alignments) y es el insight clave que hace efectivo a AlphaFold2.
Como otros sesgos inductivos, sin embargo, eso perjudica la generalización.
La apuesta por el escalamiento
Si se mira la línea de tiempo de las leyes de scaling para LLMs y la liberación de modelos de predicción de estructura, el equipo ESM duplicó su apuesta contra los MSAs después del lanzamiento de AlphaFold2. Obviamente eso requiere mucha confianza en la hipótesis del escalamiento.
ESM se desarrolló en un momento en que las leyes de scaling y la Bitter Lesson iban resultando cada vez más ciertas. El éxito salvaje de AlphaFold2 debió ser a la vez excitante y amargamente decepcionante. Pero usar MSAs significa que el modelo depende de datos de entrenamiento que contengan MSAs para ser preciso en un dominio dado. Para cosas como los anticuerpos, que no tienen MSAs en los que entrenarse, AlphaFold tiende a desempeñarse pobremente.
ESM toma un enfoque diferente: aprender la relación entre proteínas mediante entrenamiento no supervisado sobre toda la diversidad que pueda encontrar, y luego correlacionar eso con estructuras conocidas del Protein Data Bank (PDB) y otras fuentes. En otras palabras: un World Model.
¿Qué es un World Model para proteínas?
Un World Model implica usar entrenamiento no supervisado para aprender patrones abstractos de los datos:
- La abstracción debe ser semántica: las construcciones novedosas representan cosas que obedecen las reglas del mundo real.
- La abstracción debe ser composicional: recombinar patrones distintos lleva a construcciones novedosas y a menudo válidas.
- La abstracción debe soportar generalización: predice cosas en el mundo real para las que no fue entrenada.
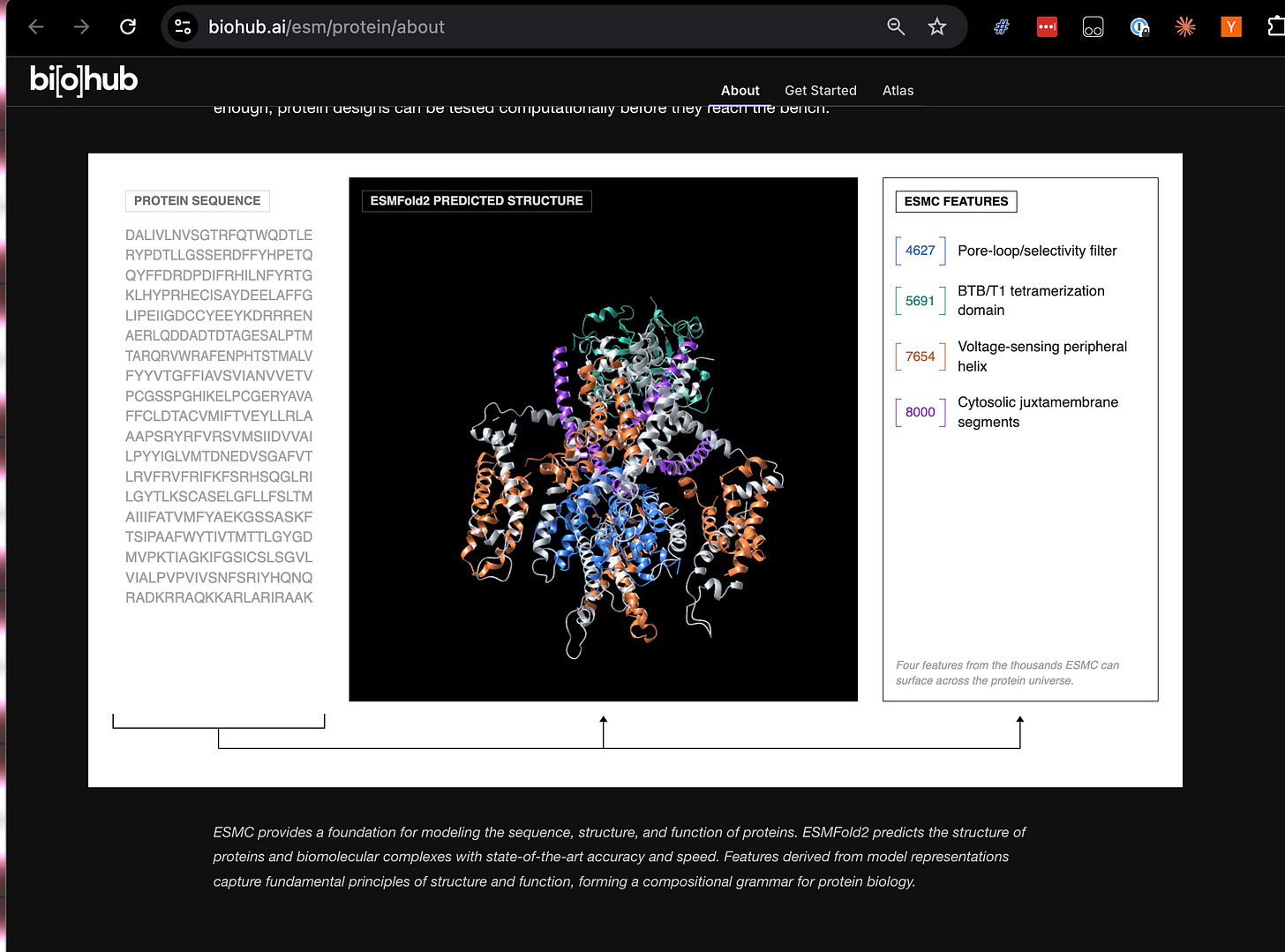
Una vez con un world model, se le pueden anclar "heads" para tareas downstream: predecir propiedades de una proteína, descomponer sus rasgos funcionales o buscar en la representación proteínas que cumplan criterios de diseño. Los dos grandes modelos que BioHub acaba de liberar bajo licencia MIT mapean directamente a este esquema:
- World model → ESMC (un modelo entrenado sobre 2.800 millones de secuencias)
- Cabeza de predicción de estructura → ESMFold2
Una manera interesante en la que el world model puede "predecir cosas" es generar secuencias de proteínas y luego medir las propiedades predichas, como afinidad de binding, en el laboratorio.
Features semánticos: la célula como computador
Sabíamos que los genes son como programas computacionales, pero la analogía suele desinflarse ahí. Una mejor analogía: el núcleo celular como dispositivo de almacenamiento, el ribosoma como JIT compiler y runtime, los features semánticos aprendidos vía Sparse Auto Encoders como funciones, las proteínas como procesos que interactúan en workflows (rutas de señalización) para producir comportamientos (fenotipos).
Como las funciones, los features SAE tienen una composición jerárquica, desde estructuras locales, secundarias y terciarias, hasta motivos conceptuales como integraciones de membrana, regiones desordenadas y enlaces disulfuro.
Lo más llamativo: el SAE dedica alrededor de 686 features (5 a 10% del presupuesto) a regiones intrínsecamente desordenadas (IDRs), que no tienen estructura para predecir. El modelo representa el desorden mismo como concepto, con sub-features para distintos sabores (poliampholyte, polar tract, prion-like).
¿Por qué importa?
El paper convierte el plegado de proteínas en un caso más del patrón ya conocido en LLMs: entrenamiento no supervisado a escala, world model emergente, heads downstream para tareas específicas. Si ESMC realmente captura biología, no solo geometría, la promesa es una biología más programable, donde diseñar una proteína se parece a componer features semánticos.